Samtools Number of Reads Linked to Multiple Contigs
- Methodology article
- Open Access
- Published:
Assessing the impact of exact reads on reducing the error rate of read mapping
BMC Bioinformatics volume xix, Article number:406 (2018) Cite this commodity
Abstruse
Groundwork
Present, according to valuable resources of high-quality genome sequences, reference-based assembly methods with high accuracy and efficiency are strongly required. Many different algorithms have been designed for mapping reads onto a genome sequence which attempt to enhance the accuracy of reconstructed genomes. In this problem, one of the challenges occurs when some reads are aligned to multiple locations due to repetitive regions in the genomes.
Results
In this newspaper, our goal is to subtract the error rate of rebuilt genomes by resolving multi-mapping reads. To reach this purpose, we reduce the search space for the reads which tin exist aligned against the genome with mismatches, insertions or deletions to decrease the probability of incorrect read mapping. We propose a pipeline divided to iii steps: ExactMapping, InExactMapping, and MergingContigs, where exact and inexact reads are aligned in 2 separate phases. We examination our pipeline on some faux and existent information sets past applying some read mappers. The results show that the two-footstep mapping of reads onto the contigs generated by a mapper such as Bowtie2, BWA and Yara is effective in improving the contigs in terms of fault charge per unit.
Conclusions
Cess results of our pipeline suggest that reducing the error rate of read mapping, not only can amend the genomes reconstructed by reference-based assembly in a reasonable running time, but can also accept an impact on improving the genomes generated by de novo associates. In fact, our pipeline produces genomes comparable to those of a multi-mapping reads resolution tool, namely MMR by decreasing the number of multi-mapping reads. Consequently, we introduce EIM as a post-processing step to genomes reconstructed past mappers.
Groundwork
The advent of next generation sequencing (NGS) technologies by greatly increasing the book of produced data, created a genomic revolution. Massive amount of information and depression cost of these technologies make it possible to determine large parts of a genome sequence in a short time. Today, biological research on any organism from viruses and bacteria to humans depends on the genome sequence information. In addition, sequences of organisms have an important part in agreement diseases.
In order to reconstruct a genome sequence based on NGS data, genome assembly, 1 of the challenging problems in bioinformatics, is divers. At that place are 2 unlike approaches to model genome assembly: de novo and reference-based associates. In the first model, a novel genome sequence is reconstructed from scratch by only applying NGS reads. In the 2d i, a reference genome is employed to gather the NGS reads by mapping them onto the reference.
Because of the large volume of NGS reads, established alignment algorithms such every bit Smith-Waterman aren't efficient for read mapping. To reduce search infinite, several algorithms have been developed [ane–five] using the seed-and-extending approach in which the reads are mapped onto the reference in two chief steps. Firstly, some subsequences of each read are selected as seeds to observe their positions in the reference. In this way, the candidate locations of the reads are determined speedily. Secondly, each read is aligned to its candidate locations past a dynamic programming algorithm in order that the actual mapping positions are obtained.
During the past years, various algorithms have been designed to improve the accuracy and efficiency of mappers [six–xiii]. Although these algorithms represent appropriate approaches to reduce the time and space complication, resolving multi-mapping reads in genome reconstruction has remained a challenge. Due to repetitive regions within the genome, some reads can be mapped to multiple locations of the reference genome. Multi-mapping reads may be aligned at incorrect locations since the read set contains sequencing errors and genetic variations relative to the reference. As a result, some errors such as mismatches and indels (insertions or deletions) are introduced to the reconstructed genome. Read mappers oft randomly select one of the locations for a multi-mapping read as the primary 1. Recently, a post-processing tool (MMR) has been developed [14] to observe optimal locations for multi-mapping reads within DNA- and RNA-seq alignment results. It resolves the problem based on the assumption of aligned reads coverage uniformity.
In this study, we innovate a new view to resolving multi-mapping reads past increasing the charge per unit of reads aligned uniquely to the reference in order to decrease the fault rate of the reconstructed genome sequence. For this aim, we separate the reads into two groups in accord with the reference genome. The idea is inspired by the following fact.
Consider a target genome (the genome from which a ready of reads is sampled) which is highly similar to the respective reference genome. If the read set is mapped onto the reference, high percentage of the reference can be covered by the reads uniquely aligned without mismatches and indels (exact reads). Leftover alignable reads (inexact reads) are so mapped to the remaining parts of the reference. Therefore, to reconstruct most of the target genome, it is enough to find the locations of reads which have unique verbal-matching with the reference. The rest of the target genome can be rebuilt by aligning remaining reads confronting the reference with mismatches and indels.
Most of the existing read mappers don't consider any differences between the mapping of exact and inexact reads. For example, hash-based mappers detect seeds which support mismatches (space-seeds) and gaps on the whole reference genome for all reads [15]. On the i mitt, consecutive seeds are plenty for exact reads and using space-seeds leads to excessive retentiveness consumption. On the other mitt, inexact reads are aligned past finding candidate locations on the whole reference genome, while according to high similarity between a target genome and its reference, searching in pocket-size parts of the reference is sufficient to find these types of reads.
Based on defining reads in ii types: exact and inexact reads, nosotros nowadays a pipeline (EIM - mapping Exact and Inexact reads separately and then Merging the constructed contigs) for resequencing of a genome. To assess our pipeline, we have chosen Bowtie2 [7] as a highly cited and user-friendly mapper and used some real and simulated read sets. For a more consummate evaluation of EIM pipeline, ii other mappers are also used. Our results illustrate that EIM pipeline improves the quality of genomes reconstructed by the mappers in terms of error rate and yields comparable results to MMR in reducing errors.
Methods
Let S=s 1 s 2…s L denote a Deoxyribonucleic acid sequence in which ∀ i≤i≤L south i ∈{A,C,G,T,N}; and |Due south| denote the length of Southward. A genome sequence is a long DNA sequence. A set of paired reads is defined as \(R=\{\langle r_{1},r^{\prime }_{ane}\rangle,\langle r_{2},r^{\prime number }_{2}\rangle,\ldots,\langle r_{m},r^{\prime number }_{one thousand}\rangle \}\) where for each i, r i and \(r^{\prime }_{i}\) are short Dna sequences with length of k.
We propose a three-step pipeline (Fig. 1) for reference-based assembly as below, where a prepare of paired reads R and a genome sequence G are given every bit inputs:
- i.
ExactMapping
Fig. 1 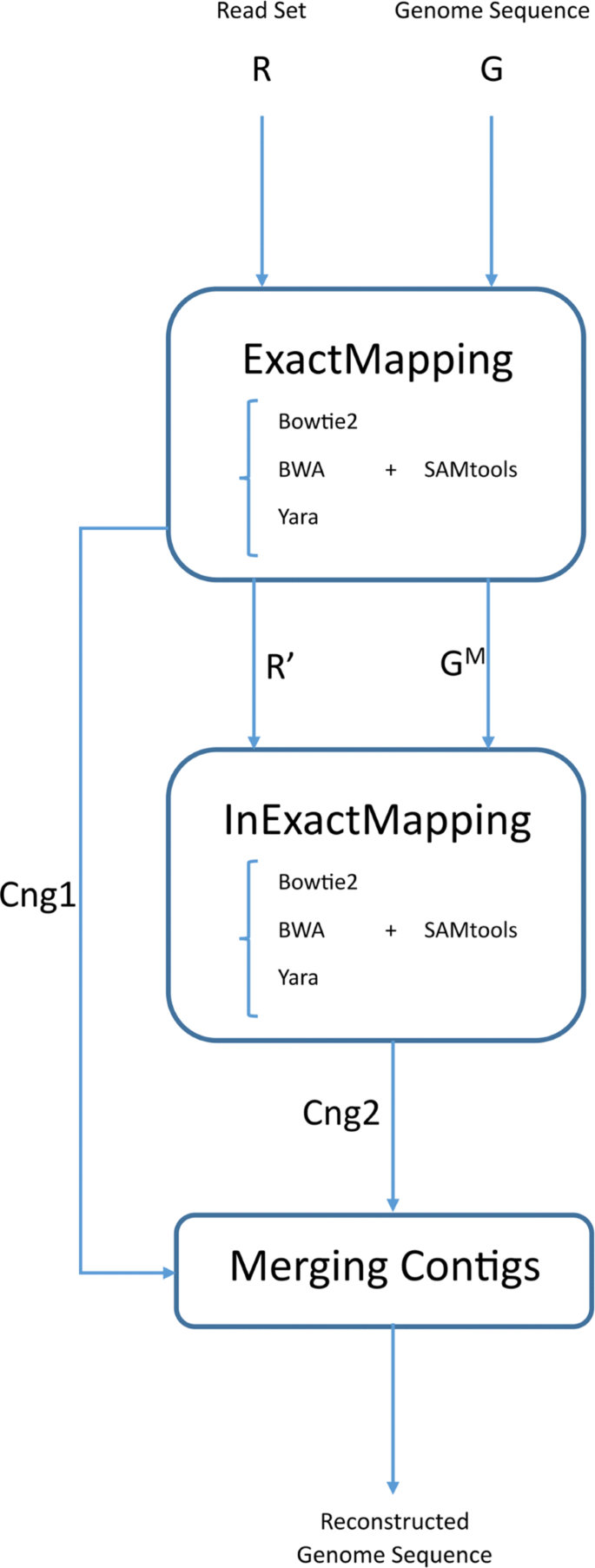
EIM pipeline overview and applied tools. The first pace has three outputs: leftover reads (R ′), modified remaining parts of the genome sequence (G Thousand ) and exact contigs (C n g1). The output of the second step is inexact contig gear up indicated past C n g2. Currently, EIM can apply 1 of mappers Bowtie2 [7], BWA [8, 9] and Yara [10] for mapping reads
The prepare of reads is mapped onto the genome sequence without mismatches and indels. Then an exact contig set chosen C n gone is generated from uniquely mapped reads.
- ii.
InExactMapping
The remaining reads from previous step are mapped onto the regions of the genome which are covered with no contigs of C northward thou1 to construct an inexact contig ready named C n g2.
- 3.
MergingContigs
The two contig sets, C n g1 and C north gii are merged to build up ultimate contigs.

In the following, each step of EIM pipeline is described in detail.
ExactMapping
In this footstep, we should utilize a mapper to align the set of reads with the genome without mismatches and indels. In this regard, the genome G and the read gear up R are given to the mapper as inputs. After running the mapper, ii outputs are produced: i) set R ′ ⊂ R containing unmapped and multi-mapping reads 2) SAM file [xvi] including the data of the alignment. And then consensus sequence C is built upwardly from uniquely mapped reads in the SAM file, where C is a Dna sequence with length |G|. Afterwards, a set of contigs called C northward g1 is generated by breaking the sequence C at each position of 'N'.
InExactMapping
In this phase, genome sequence G=g one k 2…yard n is modified based on consensus sequence C=c one c 2…c northward to generate a new genome called G Yard . To construct genome G M , the following steps are taken:
- 1:
Make sequence \( C^{\prime number }= c^{\prime }_{1}c^{\prime number }_{2} \ldots c^{\prime }_{n} \) as follows:
$$c^{\prime number}_{i} =\left\{ \begin{array}{ll} N & c_{i} \in \{ A,C,G,T\}, \\ g_{i} & c_{i}=N, \end{array}\right. $$
where C ′ contains all parts of genome Yard covered with no contigs of C due north gi.
- 2:
Generate sequence \( One thousand^{M}= one thousand^{M}_{1}thousand^{Thousand}_{two}\ldots one thousand^{Chiliad}_{n} \) by extending each face-to-face nucleic acrid sequence as:
$$yard^{M}_{i} =\left\{ \brainstorm{array}{ll} c^{\prime number}_{i} & c^{\prime}_{i} \in \{A,C,G,T\}, \\ g_{i} & c^{\prime}_{i}=North \& \exists_{j=1}^{k} c^{\prime}_{i \pm j} \in \{A,C,G,T\}, \\ N & o.due west, \end{array}\right. $$
where k is equal to the read length.
Then G M is broken at each position of 'N', and every bit a result a set of contigs is obtained. Later that, a mapper is used in order to align R ′ confronting the gear up of contigs with mismatches and indels. Finally, a consensus sequence is made from mapped reads in the SAM file for each contig and added to C n g2.
MergingContigs
In this function, the two contig sets C n g1 and C n k2 generated respectively at the steps of ExactMapping and InExactMapping, are combined to rebuild the target genome. Although C n m1 contains large contigs which make upwardly most of the target genome, C n 10002 is required to produce larger contigs including the differences with genome G. We merge the contig sets without alignment because the positions of contigs relative to the genome G are known. In this fashion, every two contigs of C n ki are joined past a contig of C n g2 overlapping with both of them. Merging method is described in more than detail below.
The union of C n thousandi and C north chiliad2 contig sets is defined equally \(Cng=\left \{\prec D^{i},south^{i},e^{i},t^{i} \succ \mid D^{i}=d^{i}_{1}d^{i}_{ii}\cdots d^{i}_{due east^{i}-s^{i}+ane}\right \}\) where for each i, D i is a contig belonging to either C n g1 or C north g2. The beginning and end positions of contig D i on the reference are shown by due south i and e i , repectively. It should be noted that s i <s i+1 and e i <east i+1. Moreover, the value of t i is set to 1 (or 2) when D i ∈ C n g1 (or D i ∈ C n g2). In the post-obit, all the contigs in Cng are merged past a recursive equation:
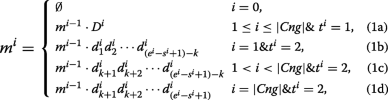
where k is equal to the length of a read, and |C northward g| is the number of contigs in the Cng set. For each i, g i denotes the merged sequence achieved past combining D one to D i . Function (1a) of the above equation shows that each contig of C north one thousand1 has to be completely inserted to the merged sequence every bit it is highly probable that the contig has been made correctly. Parts (1b), (1c) and (1d) indicate how to insert a contig of C n g2 to the merged sequence after removing the extended parts (with length of 1000). The ultimate merged sequence is represented by m |C n g| which may include some Ns because of C north yard2 contigs. Thus m |C n g| sequence is broken at each position of 'N' for generating output contigs of EIM pipeline.
Datasets
Several real and false datasets are used to evaluate the accurateness of EIM pipeline. The first real dataset is an Illumina MiSeq pair-cease read set from E. coli downloaded from [17, xviii] which consists of about 1.5 million paired reads of 151 base-pair (bp) with coverage depth 100×. We apply Escherichia coli str. K12 substr. MG1655 [GenBank:NC_000913] as a reference genome and Escherichia coli O145:H28 str. RM12581 [GenBank: CP007136.ane] as a related strain.
The second dataset includes four homo chromosome read sets: Chr1, Chr10, Chr14 and Chr21 extracted from samples. The whole human genome samples are downloaded from the SRA database of National Center for Biotechnology Information (NCBI) with accession numbers SRR67780, SRR67785, SRR67787, SRR67789, SRR67791, SRR67792, SRR67793. The homo reference genome GRCh38 is downloaded from [xix]. All read sets contain 101 bp paired reads with the properties shown in Table ane.
We simulate several read sets for a prokaryotic and eukaryotic genome: E. coli and Arabidopsis thaliana. To simulate reads for Due east. coli, we create four genome sequences, Due east. coli-Mut1 to Eastward. coli-Mut4 derived from Eastward. coli K12. So Illumina read sets, ReadSet1 to ReadSet8 and ReadSet9 to ReadSet12 are simulated for mutated genomes by DWGSIM [20] and ART [21], respectively. E. coli-Mut1 and Eastward. coli-Mut2 have unmarried nucleotide variants (SNVs) with the rate of 0.1%. Due east. coli-Mut2 has SNVs of random size among 1 to 3. E. coli-Mut3 has SNVs and deletions of the rates 0.09% and 0.01% respectively. E. coli-Mut4 has SNVs and insertions of the rates 0.09% and 0.01% respectively. The read sets, ReadSet1 to ReadSet4 are simulated such that the length and coverage depth of the reads are similar to those of the real read set from East. coli K12 genome (i. e. more than i.five 1000000 paired reads of 150 bp). The read sets, ReadSet5 to ReadSet12 are faux with low coverage (i. e. nearly 3000 paired reads of 150 bp) and sequencing error. The properties of faux reads are shown in Table 2.
To generate reads for Arabidopsis thaliana, nosotros create a genome sequence derived from TAIR10 [GenBank: CP002684.1-CP002688.1] reference genome. Firstly, TAIR10 genome sequence is mutated based on bur-0 strain variations obtaining from [22]. So an Illumina read set including 15.vi one thousand thousand paired reads of 150 bp with coverage depth of 20× is faux by ART simulator.
Tools
Some tools are utilized for running EIM pipeline equally follows. We use DWGSIM [xx] and ART [21] for simulating reads, Bowtie2, Yara [10] and BWA [eight, 9] for mapping reads, and SAMtools [16] for making consensus sequences. We likewise implement a simple hash-based aligner called ExactMapper for mapping reads without mismatches and gaps to make the pipeline faster.
The assessments on big genomes including man chromosomes and Arabidopsis thaliana are performed on a desktop which has a 3.60GHz Intel(R) Core(TM) i7−6850K six-core processor and 32GB of RAM running 64-bit Ubuntu 18.04 LTS. The other assessments are performed on a laptop with an Intel(R) Cadre(TM) i7−3517U processor and 8GB of RAM running 64-bit Ubuntu 15.x.
At ExactMapping pace, we apply ExactMapper aligner for small genomes to generate a SAM file and excerpt remaining reads (unmapped and multi-mapping reads) simultaneously. Next, SAM file is given to a script to build upward a consensus sequence C from uniquely mapped reads. At InExactMapping stride, we employ one of the same mappers with advisable parameters and then construct the consensus sequence by SAMtools. For this purpose, The '--go on-masked-ref' parameter is prepare for 'bcftools call' command of SAMtools to be able to make consensus in IUPAC positions of the reference genome.
It should be noted, for big genomes such as the man chromosome 14, nosotros use Bowtie2 in ExactMapping step. The '--score-min' parameter of Bowtie2 is set to the value ' C,0,−i' to only map the reads with exact matches to the genome. Th unmapped and multi-mapping reads are extracted from the SAM file by a script and the consensus sequence is constructed by SAMtools.
Evaluation metrics
To evaluate EIM pipeline, we calculate some contiguity and quality metrics by QUAST [23] for contig sets (genomes) reconstructed past ExactMapping stride, EIM and the mappers.
We apply two metrics to compare the contiguity of the contig sets every bit follows:
-
Contigs-500: The number of contigs with length of greater than 500 bp belonging to the contig set.
-
N50: The length of the smallest contig in the set that contains the fewest (largest) contigs whose combined length represents at to the lowest degree 50% of associates [24].
We use quality metrics for indicating the accuracy of the reconstructed genomes. To calculate some quality metrics, each set of contigs is aligned to the target (or reference) genome to discover the number of errors regarding to each contig prepare equally below:
-
Errors: The total number of mismatches and indels (insertions and deletions) in the aligned contigs relative to the target genome.
-
IUPAC-codes: The total number of IUPAC ambivalence positions in the contig set.
-
Genome-Fraction: The percentage of the target (or reference) genome covered past the aligned contigs.
when the target genome is not available, we use the following quality measure to test the accuracy of the reconstructed genomes.
-
Remapped-Reads: The percentage of the reads which are identically mapped (i.eastward. without mismatches and indels) onto the contigs.
Results
A set of reads and a reference genome are given to EIM pipeline as inputs and so EIM constructs a ready of contigs every bit output by stepwise mapping of the reads onto the reference. The sequencing errors and genetic differences as well as repetitive regions in the genome are the factors which introduce mapping errors such as mismatches and indels into the contigs relative to the target genome.
To evaluate the results of EIM pipeline, we apply different datasets in terms of similarity between the target and reference genomes as follows:
- 1.
By considering a reference genome identical to the target genome, we initially assess our pipeline where the existent read fix from E. coli K12 includes sequencing errors.
- 2.
According to the loftier similarity betwixt whatsoever human genome and the human reference, nosotros investigate results of EIM pipeline where the real reads from a man chromosome 14 comprise sequencing errors equally well every bit SNVs. Information technology is to be noted that the target genome is non available.
- 3.
Past simulating some target genomes highly similar to East. coli G12 genome, we examine EIM pipeline in which the simulated reads include SNVs. In this mode, we can exam the accurateness of EIM more precisely since the target genomes are available.
- four.
By using a closely related genome to E. coli K12 equally a reference, nosotros perform EIM pipeline on a real read set from Eastward. coli K12 to assess our pipeline where the similarity betwixt the target and reference genomes is not very high.
For completing the evaluation of EIM, we apply unlike mappers on a real read gear up from E. coli M12 and a closely related genome to information technology as a reference, and so compare the results of EIM pipeline to the respective mappers. In addition, we evaluate our pipeline on eukaryotic genomes of human and Arabidopsis thaliana.
Assessment of EIM on a real dataset of E. coli K12
To test the accuracy of EIM, we examine the outcome of sequencing errors without considering any other factors. For this purpose, E. coli K12 genome and its reads generated by using Illumina are given to EIM as inputs. Accordingly, the target and reference genomes are the same and the read set up includes sequencing errors.
An Illumina sequencer has an error rate of <0.1% [25], because of which only 61.79% of the reads tin can be mapped at the get-go step of EIM pipeline (ExactMapping). All the same, contigs constructed from the uniquely mapped reads cover nearly entire of the target genome (99.995% in Table 3). At the 2d step of our pipeline (InExactMapping), remaining reads from the first step are mapped onto just 0.005% of the reference. As shown in Tabular array 3, the last footstep of EIM (MergingContigs) produces a face-to-face contig including 2 errors, while Bowtie2 mapper makes 11 contigs containing the same number of errors on this sample data. Although Bowtie2 generates more contigs than EIM, the Genome-Fraction values of both contig sets are the aforementioned (100%) because the gaps between contigs of Bowtie2 are too small compared to the total length of the target genome.
This cess shows that contig sets reconstructed by EIM and Bowite2 are the aforementioned in terms of accuracy when the read set contains sequencing errors.
Assessment of EIM on a real dataset of human chromosome 14
In this assessment, our goal is to investigate the accuracy of EIM where the prepare of reads extracted from a genome includes sequencing errors also equally SNVs and indels relative to the reference. We perform EIM on the human chromosome 14 reference and the reads from a homo chromosome 14.
Due to sequencing errors and genetic differences between human genomes, only about one-half of reads (58.33%) are aligned at the ExactMapping step. The contigs constructed from this volume of the reads comprehend 93.22% of the chromosome 14 reference (Table 3). Furthermore, the results presented in Table 3 testify that EIM makes significantly fewer contigs than Bowtie2. In other words, the comparison of N50 values indicates that EIM tin make a contig ready more contiguous than that of Bowtie2. Moreover, the contigs of EIM include fewer errors relative to the reference than those of Bowtie2. Although comparing with the reference genome gives insight into the error charge per unit of the reconstructed genomes, some differences are truthful differences rather than errors. Since the target genome is non available, we use the read gear up to assess the accuracy of EIM. In this mode, the reads are mapped without mismatches and indels to the reconstructed genomes to calculate Remapped-Reads values. The results of the remapping show that the Remapped-Reads values for the genomes reconstructed past EIM and Bowtie2 are 60.87% and 58.68% respectively. This is an appropriate evidence that the reconstructed genome by EIM is more accurate than that of Bowtie2.
Our results prove that when the target and reference genomes are highly like, EIM pipeline can reconstruct a more accurate genome than the one rebuilt past Bowtie2 mapper.
Assessment of EIM on simulated information
To assess the accuracy of EIM more precisely, the target genome sequences are required. Since target sequences are typically not bachelor for about of individuals and strains, we use false data. To do so, nosotros make some genome sequences derived from Due east. coli K12 genome by creating mismatches and indels using different rates and then simulate read sets from the mutated genomes (Table 2).
We test EIM pipeline on ReadSet1 (Table 2) and Eastward. coli Thou12 as a reference genome. To compare contigs generated past EIM and Bowtie2, we align both contig sets against E. coli-Mut1 (the target genome) and present the results in the second, third and last columns of Table iv. Although EIM pipeline rebuilds a face-to-face contig, information technology introduces more errors than Bowtie2. It is also worth mentioning that the contigs of ExactMapping footstep of EIM called Exact contigs have 90.285% Genome-Fraction value which in comparison with that obtained by real data experiment (99.995% in Table 3) is very low. It seems that a lower Genome-Fraction value of Verbal contigs leads to the college errors in the last contigs produced by EIM.
Nosotros need to signal out that the more than fraction of the target genome is covered by Exact contigs, the smaller parts of the reference remain for InExactMapping step of EIM. Hence the probability that the leftover reads are aligned at true locations is increased and as a result, the error rate of the reconstructed genome is reduced. Furthermore, the fraction of the target genome covered by Exact contigs is directly proportional to the similarity betwixt the target and reference genomes. In other words, the college similarity between the target and reference genomes leads to fewer errors in the genome reconstructed by EIM pipeline. Accordingly, since the genome sequence reconstructed past a mapper is more similar to the target genome than to the reference (ref), the genome sequence reconstructed past Bowtie2 (cns-bt) is fed to EIM instead of the reference as input.
The results can be seen in the 4th and fifth columns of Tabular array 4. The comparing of the second and fourth columns shows that by giving the genome sequence reconstructed past Bowtie2 instead of the reference sequence to EIM as the input, the Genome-Fraction value of Exact contigs increases from 90 to >99%. In addition, the number of errors in last contigs of EIM decreases from 45 to 28. It suggests that the genome sequence reconstructed by a mapper is a meliorate input for our pipeline as it leads to a lower error rate. Our analysis up to this signal shows that by feeding cns-bt instead of ref to EIM pipeline as input, the mistake rate is reduced. Information technology is important to annotation that the error rate decreasing is valuable only when EIM rather maintains the same Northward50 and Genome-Fraction values as those of the input genome. Even so, the results of EIM in the fifth column compared to the last cavalcade of Table 4 indicate that this condition is not satisfied.
We observed that cns-bt includes 137 IUPAC-codes while ref contains no IUPAC-codes. Furthermore, the genome reconstructed by mapping a read fix onto a reference sequence containing IUPAC-codes is less contiguous than the reference because SAMtools makes a consensus sequence including 'Due north' in the IUPAC-code positions. Thus the being of IUPAC-codes in the input genome of EIM yields a more fragmented genome as output. To solve this result, we execute SAMtools with a parameter assuasive to build consensus in the IUPAC-lawmaking positions instead of substituting 'N' ambiguity character ("Tools" subsection). Every bit shown in the 6th column of Table four, EIM with this modification makes contigs which in addition to including less errors than cns-bt (the input genome), are nearly as face-to-face as cns-bt and with high coverage of the target genome. In the following, EIM described in the fifth and sixth columns of Table four are considered equally versions one (51) and two (v2), respectively.
Tables 5 and 6 represent the results of applying EIM (v2) pipeline and Bowtie2 mapper to the simulated read sets with high and low coverage, respectively. As illustrated by the results, non just tin EIM (v2) decrease the error and IUPAC-lawmaking rates, but it tin can besides maintain the contiguity and Genome-Fraction value very shut to Bowtie2.
The results of this assessment prove that our pipeline tin improve the genome sequence reconstructed past Bowtie2 mapper in terms of accuracy when a highly similar reference to the target genome is available and the read set includes SNVs relative to the reference.
Cess of EIM on a real dataset of E. coli K12 and a closely related genome
In this assessment, we examine the accuracy of EIM when similarity between the target and reference genomes is not so loftier. The awarding is where a reference is not bachelor and a closely related genome is used as a reference. We utilize Eastward. coli O145:H28 as a closely related genome to E. coli K12.
To evaluate EIM on the read set from E. coli K12, a genome sequence is reconstructed from mapping the reads onto E. coli O145:H28 genome by Bowtie2, then the reconstructed genome and the reads are given to EIM as inputs. Table 7 shows that the contig sets generated by EIM (vone) and EIM (52) incorporate fewer errors and IUPAC-codes than that of Bowtie2. Moreover, EIM (v2) can make contigs which have nearly the same Genome-Fraction value and N50 size as those of Bowtie2.
It should be noticed that the Genome-Fraction values of the contigs produced by EIM and Bowtie2 are less than 90%. In such cases where there is no reference available and the related genome is not highly similar to the target genome, de novo genome assembly is a better approach for reconstructing the genome sequence. Nonetheless, the genome sequences generated by de novo assemblers are not mistake-free. For this reason, approaches for improving the accuracy of de novo assembled contigs are needed. Hither we utilize the contigs generated by EIM to improve the contigs produced by a de novo assembler. In fact, we utilise version one of EIM pipeline because contigs of EIM (fivei) include less errors than those of EIM (v2). The read set is assembled by MaSuRCA [26], 1 of the all-time assemblers at GAGE-B [27], so the contigs constructed by EIM and MaSuRCA are combined into a contig fix including fewer errors than the contigs of MaSuRCA (Tabular array vii).
This analysis indicates that when a closely related genome is used as a reference, and thus the reference and target genomes are non highly similar, EIM (v2) can reconstruct a genome sequence with the same contiguity and Genome-Fraction value including less errors and IUPAC-codes than the genome reconstructed by Bowtie2 mapper. In addition, the genome rebuilt past EIM (vone) can decrease the error charge per unit of a genome sequence generated past a de novo assembler such as MaSuRCA.
Evaluation of EIM by different mappers
To evaluate the performance of our pipeline by using mappers other than Bowtie2, nosotros select BWA as a pop and widely used mapper and Yara as one of the state-of-the-art mappers. We employ the 3 mappers and version ii of EIM on the read set from E. coli K12 and Eastward. coli O145:H28 genome every bit a reference. For each mapper, the genome reconstructed by the mapper is given to EIM (5two) as input and the mapper itself is practical for adjustment reads in the 2d pace of EIM (v2) (i. e. InExactMapping).
As illustrated in Fig. two, for all mappers, EIM pipeline maintains N50 size and Genome-Fraction value close but non identical to those of the mappers (Fig. 2a and b). It as well reduces the number of errors and significantly decreases the number of IUPAC-codes (Fig. 2c and d).

The comparison of contigs generated by Bowtie2, Yara and BWA with the respective contigs of EIM on the real read set of E. coli K12. Firstly, the mappers were executed on the read gear up and the reference, then the contig sets were generated. Secondly, for each mapper, EIM (v2) was run on the read prepare and the contig set synthetic past the mapper while using it at the second step for mapping. Finally, the contiguity and quality of contigs were computed as a N50 size b Genome-Fraction value c The number of errors d The number of IUPAC codes. In addition, the running time of obtaining contigs was measured and showed in seconds (due east)
Effigy 2e shows the running times of the 3 mappers compared to EIM. Since the input genome of EIM is congenital by a mapper, the running time of reconstructing a genome by EIM is the total of mapper and EIM pipeline runtimes. In add-on, the running time of reconstructing a genome by a mapper is the total of read mapping and consensus amalgam runtimes, which the second one is more time-consuming. Our pipeline decreases the computational time of making a consensus by a ii-step mapping. In ExactMapping, most of the reads are exactly aligned and a SAM file is fabricated from which the consensus sequence tin exist constructed past a simple and fast script without using SAMtools. Moreover, only a low percent of reads is transferred to InExactMapping step and thus the consensus sequence is made apace past SAMtools in this stage. Consequently, the overhead time of reconstructing the Eastward. coli genome by EIM pipeline afterward running a mapper is less than ane-tertiary of that of the respective mapper (Fig. 2e).
This evaluation demonstrates that EIM pipeline tin be used as a post-processing tool to better the genome reconstructed by a mapper to a more accurate i in an acceptable runtime while maintaining the contiguity and Genome-Fraction value of the input genome.
Evaluation of EIM on de novo assembled genomes
In this department, we appraise the effect of EIM pipeline on the results of de novo assemblies. For this purpose, we compare EIM with Pilon framework [28] and Columbus module of Velvet assembler [29]. These tools get a draft or reference genome and mapped reads on it, to utilize read mappings for improving genome assembly.
In the following, we first generate 2 genomes by Velvet and MaSuRCA assemblers on the real read set from E. coli K12. Then each draft genome is inputted to EIM, Pilon, and Columbus.
Every bit illustrated in Fig. iii, all frameworks reduce the number of errors and dramatically decrease the number of IUPAC-codes when that of the typhoon genome is besides high (Fig. 3a and b). Although EIM and Columbus decrease N50 size (Fig. 3c), they maintain Genome-Fraction value close to those of typhoon genomes (Fig. 3d).

The comparison of contigs generated by EIM, Pilon and Columbus on the real read prepare of E. coli K12.Firstly, two draft genomes were generated by Velvet and MaSuRCA de novo assemblers. Secondly, EIM, Pilon and Columbus were run by each typhoon and mapped reads on the draft. Finally, the quality and contiguity of contigs were computed as a The number of errors b The number of IUPAC codes c N50 size d Genome-Fraction value
The results of this comparison show that EIM pipeline has an bear upon on reducing the mistake rate of the genomes generated by de novo associates.
Evaluation of EIM on eukaryotic genomes
For the final evaluation, we run EIM pipeline on the datasets of human as a mammalian and Arabidopsis thaliana equally a model establish. To evaluate EIM on homo, we select the smallest and the largest chromosomes as well as a chromosome with average length namely Chr21, Chr1, and Chr10, respectively and extract the reads of each one from existent samples of the whole homo genome. Then we run EIM on each dataset separately. For evaluating our pipeline on Arabidopsis thaliana, we simulate a dataset for all chromosomes of bur-0 strain and use TAIR10 equally the reference to run EIM.
Equally shown in Table eight, EIM pipeline reduces error rates on all iii man chromosomes and bur-0 strain of Arabidopsis. To exist precisely measured the accuracy of generated contigs, the reads are exactly mapped onto each contig set to calculate Remapped-Reads value. As seen, EIM increases Remapped-Reads values. Furthermore, the results show that our pipeline considerably increases the N50 size of contig sets generated for homo chromosomes because of the high similarity between human genomes.
In lodge to examine the outcome of dissimilar chromosomal regions on accuracy of EIM, we test our pipeline on portions of a human chromosome. To achieve this goal, we divide Chr1, the largest human chromosome, to twenty five aforementioned-length regions as follows:
$$P = \{p_{i}, \ldots, p_{25}\} \;\; for \;\; each \;\; i \;\; |p_{i}| \simeq 10 Mbp. $$
The number of ambiguity characters (Ns) is assessed in each p i 1≤i≤25 (Fig. 4). Nosotros omit p xiv considering this region is a whole sequence of Ns. Nosotros and so run EIM on the read set up of Chr1 and each p i 1≤i≤25a north d i≠14, separately.

The distribution of N characters in the regions of Chr1. The regions of p 14, p 13 and p 15 take 54%, 24% and 20% of N characters of Chr1, respectively. The centromere consists of p 13, p 14 and p xv regions which contain 97% Ns of Chr1
As shown in Fig. 5, EIM pipeline increases N50 values and reduces fault numbers, and significantly decreases IUPAC numbers for all regions. Note that, because of the high fraction of Ns in centromere region, contigs generated by Bowtie2 and EIM on p 13 and p fifteen have low N50 size and low error numbers (Fig. 5a and d).

The comparison of contigs generated past Bowtie2 and EIM on the regions of Chr1. Firstly, Chr1 was divided to some regions, p i 1≤i≤25. Secondly, Bowtie2 and EIM were run past the read set of Chr1 and each p i ane≤i≤25a n d i≠14, separately. Finally, the quality and contiguity of contigs were computed every bit a N50 size b The Remapped-Reads value c The number of IUPAC codes d The number of errors
In addition, EIM increases Remapped-Reads values for all regions except for the first one (Fig. 5b). To explore the reason, nosotros pause the p i region from Ns and select ii of five yielded portions chosen \(p_{1\_1}\) (∼2.1 Mbp) and \(p_{one\_2}\) (∼7.2 Mbp) for assay considering their length is more than i Mbp. Then we run EIM on the read set of Chr1 and \(p_{ane\_1}\) and \(p_{1\_2}\) regions, separately. The results prove that the Remapped-Reads value of contigs generated by EIM is 0.05% more than than that of Bowtie2 for \(p_{1\_2}\) while this value is 0.35% less than that of Bowtie2 for \(p_{i\_1}\). Thus the shorter portion i.e. \(p_{ane\_1}\) leads to decreasing of the Remapped-Reads value of p 1 region. According to this observation, we examine GC-content of all regions of Chr1. The GC-content of \(p_{1\_1}\) is 56% while GC-content of \(p_{1\_2}\) and other regions are less than 50% (Fig. 6).

GC-content of contig sets generated by EIM on the regions of Chr1. As shown, \(p_{1\_1}\) has the maximum GC-content among all regions
The results from GC-content analysis suggest that running EIM on genomic regions with less than 50% GC-content tin generate contigs which are more accurate than those of a mapper.
Discussion
Every bit mentioned in the "Background" section, one of the most challenging aspects of genome sequence reconstruction from NGS data is the being of multi-mapping reads. We merits that EIM pipeline decreases the number of multi-mapping reads and thus reduces the error rate of the reconstructed genome. To demonstrate this claim, nosotros analyse each step of EIM separately. Let the input genome sequence of EIM be the genome reconstructed by a mapper like Bowtie2.
At ExactMapping step, the consensus sequence is built from the reads uniquely mapped and thus the resulting Verbal contigs contain very low errors (see the 'Exact' column in Tables 6 and 7). Therefore the number of errors in the contigs of the next step plays a determining role in the error rate of the genome reconstructed by EIM. The reads non applied in this footstep, namely multi-mapping and unmapped reads are transferred to the second step to be aligned with mismatches and indels.
At InExactMapping pace, the remaining reads are aligned to the parts of the input genome not covered by any Exact contigs so the consensus sequence is generated. To examine the effect of EIM on multi-mapping reads, we should compare the number of multi-mapping reads in this step to that obtained past mapping the reads onto the whole input genome. To do so, the reads that tin be mapped at the second stride of EIM, are aligned again to the whole input genome. Figure 7 shows that our pipeline leads to less multi-mapping reads on the simulated and real datasets. In fact, on the imitation datasets, EIM can decrease the number of multi-mapping reads by finding unique mapping locations for 17% of them on average.

Multi-mapping reads on the whole and the remaining parts of the genome. A real and 4 fake datasets were used. The orange and yellow bars show the percent of multi-mapping reads where the reads were aligned against the whole genome, and in which the reads were mapped onto the regions not covered past the contigs of the first footstep of EIM, respectively
To complete the exam of the consequence of EIM pipeline on multi-mapping reads, EIM is compared to a multi-mapping reads resolution tool, MMR. We compare the genome reconstructed by EIM to the genome obtained based on the results of MMR on the read prepare from E. coli K12 and Due east. coli O145:H28 as a reference. In this fashion, firstly, the reads are mapped by Bowtie2 onto the reference and a SAM file is generated. Then a sorted BAM file and a consensus sequence are built from SAM file every bit the inputs of MMR and EIM, respectively. MMR produces a BAM file that assigns an optimal mapping location to each multi-mapping read, while EIM generates a contig fix such that the number of multi-mapping reads are decreased.
Equally shown in Table 9, both approaches maintain the contiguity and reduce the error rate of the input. In addition, EIM tin can impressively decrease the number of IUPAC-codes from 280 to 56. The running time of reconstructing E. coli genome by EIM (330 sec) is significantly less than that of MMR (999 sec) without considering the running time of making the inputs. Notation that for reconstructing a genome based on MMR results, a consensus structure phase is required after applying MMR which causes to increase the runtime.
As shown by this analysis, the results of EIM pipeline are comparable to a multi-mapping reads resolution tool in terms of the main goal, that is, reducing the error rate of the genome reconstructed by a mapper.
Determination
The goal of our work is to improve the accuracy of contigs generated using NGS read mappers by decreasing their error rate. To attain this purpose, we design EIM pipeline which aligns the verbal and inexact reads against the genome sequence at two dissever steps to map the inexact reads more precisely. The assessment of our pipeline on imitation and real read sets testify that the separation of reads is effective in reducing the number of mismatch and indel errors with regard to the target genome and significantly decreases the number of IUPAC-codes in the input genome. The evaluation of EIM by iii mappers namely Bowtie2, BWA and Yara as well indicates that our pipeline, as a mail service-processing step to dissimilar mappers, tin ameliorate the genome sequences reconstructed by them in an adequate running fourth dimension. In improver, EIM pipeline tin can reconstruct a comparable genome to that of MMR (a multi-mapping reads resolution tool) in terms of error rate.
Abbreviations
- bp:
-
Base-pair
- BAM:
-
Binary Alignment/Map
- Chr:
-
chromosome
- cns-bt:
-
Genome sequence reconstructed by Bowtie2
- EIM:
-
mapping Verbal and Inexact reads separately so Merging the constructed contigs
- indel:
-
Insertion or deletion
- kbp:
-
Kilo base-pair
- NCBI:
-
National Center for Biotechnology Information
- NGS:
-
Next generation sequencing
- ref:
-
Reference
- SAM:
-
Sequence Alignment/Map
- SNV:
-
Unmarried nucleotide variant
- SRA:
-
Short read annal
References
-
Kent WJ. BLAT–the Smash-similar alignment tool. Genome Res. 2002; 12:656–64.
-
Li H, Ruan J, Durbin R. Mapping brusk Dna sequencing reads and calling variants using mapping quality scores. Genome Res. 2008; 18(11):1851–viii.
-
Lin H, Zhang Z, Zhang MQ, Ma B, Liy M. ZOOM! zillions of oligos mapped. Bioinformatics. 2008; 24(21):2431–7.
-
Homer North, Merriman B, South.F. N. BFAST: An Alignment Tool for Large Scale Genome Resequencing. PLoS 1. 2009; 14(11):e7767.
-
Li R, Li Y, Kristiansen M, Wang J. Soap: short oligonucleotide alignment plan. Bioinformatics. 2008; 24(5):713–4.
-
Langmead B, Trapnell C, Pop M, Salzberg Southward. Ultrafast and memory-efficient alignment of short Dna sequences to the human genome. Genome Biol. 2009; 10(3):25.
-
Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012; 9(4):357–9.
-
Li H, Durbin R. Fast and authentic short read alignment with burrows-wheeler transform. Bioinformatics. 2009; 25(14):1754–60.
-
Li H, Durbin R. Fast and authentic long-read alignment with burrows-wheeler transform. Bioinformatics. 2010; 26(v):589–95.
-
Siragusa East, Weese D, Reinert Thousand. Fast and authentic read mapping with guess seeds and multiple backtracking. Nucleic Acids Res. 2013; 41(seven):78.
-
Li R, Yu C, Li Y, Lam T-Westward, Yiu Southward-M, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009; 25(15):1966–7.
-
Gontarz P, Berger J, Wong C. SRmapper: a fast and sensitive genome-hashing alignment tool. Bioinformatics. 2013; 29(3):316–21.
-
Lee W, Stromberg Yard, Ward A, Stewart C, Garrison Eastward, Marth Grand. MOSAIK: a hash-based algorithm for accurate adjacent-generation sequencing read mapping. PLoS One. 2014; 9(3):e90581.
-
Kahles A, Behr J, Rätsch G. MMR: a tool for read multi-mapper resolution. Bioinformatics. 2016; 32(v):770–2.
-
Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Cursory Bioinform. 2010; 11(5):473–83.
-
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer Northward, Marth G, Abecasis One thousand, Durbin R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics. 2009; 25(16):2078–9.
-
Koren S, Harhay GP, Smith TP, Bono JL, Harhay DM, Mcvey SD, Bergmen NH, Phillippy AM. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol. 2013; 14(9):101.
-
PacBio Corrected Reads (PBcR) Pipeline. http://www.cbcb.umd.edu/software/PBcR/closure/index.html. Accessed 23 Oct 2018.
-
UCSC Genome Browser. http://hgdownload.soe.ucsc.edu/goldenPath/hg38/chromosomes/. Accessed 23 Oct 2018.
-
DWGSIM. https://github.com/nh13/DWGSIM. Accessed 23 Oct 2018.
-
Huang W, Li L, Myers JR, Marth GT. Fine art: a next-generation sequencing read simulator. Bioinformatics. 2012; 28(4):593–4.
-
nineteen Genomes of Arabidopsis Thaliana. http://mtweb.cs.ucl.ac.great britain/mus/www/19genomes/variants.SDI/bur_0.v7c.sdi. Accessed 23 Oct 2018.
-
Gurevich A, Saveliev Five, Vyahhi N, Tesler G. Quast: quality assessment tool for genome assemblies. Bioinformatics. 2013; 29(eight):1072–v.
-
Miller JR, Koren S, Sutton Thousand. Assembly algorithms for next-generation sequencing data. Genomics. 2010; 95(half-dozen):315–27.
-
Laehnemann D, Borkhardt A, McHardy AC. Denoising Dna deep sequencing data-high-throughput sequencing errors and their correction. Brief Bioinform. 2015; 17(1):154–79.
-
Zimin A, Marçais Chiliad, Puiu D, Roberts G, Salzberg S, Yorke J. The MaSuRCA genome assembler. Bioinformatics. 2013; 29(21):2669–77.
-
Magoc T, Pabinger S, Canzar S, Liu X, Su Q, Puiu D, Tallon LJ, Salzberg SL. GAGE-B: An Evaluation of Genome Assemblers for Bacterial Organisms. Bioinformatics. 2013; 29(fourteen):1718–25.
-
Walker BJ, Abeel T, Shea T, Priest Yard, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE. 2014; ix(11):e112963.
-
Zerbino DR, Birney E. Velvet: Algorithms for de Novo Brusk Read Associates Using de Bruijn Graphsr. Genome Res. 2008; eighteen(5):821–9.
Acknowledgements
Not applicable.
Funding
This enquiry is supported in part by a grant (No. BS-1397-01-02) from the Institute for Enquiry in Fundamental Sciences (IPM), Tehran, Iran.
Availability of data and materials
The Illumina MiSeq pair-end read set from East. coli is avialable from [17, 18]. Escherichia coli str. K12 substr. MG1655 and Escherichia coli O145:H28 str. RM12581 are available from GenBank under the accessions NC_000913 and CP007136.i-CP007136.three, respectively. The whole human genome samples are available from the SRA database of NCBI with accession numbers SRR67780, SRR67785, SRR67787, SRR67789, SRR67791, SRR67792, SRR67793. The human reference genome GRCh38 is available from[xix]. TAIR10 is available from GenBank nether accessions CP002684.1-CP002688.1. Bur-0 strain of Arabidopsis variations respective to TAIR10 reference are bachelor from [22]. EIM pipeline and simulated datasets generated and analysed during the current study are available at http://bioinformatics.aut.ac.ir/EIM/.
Writer information
Affiliations
Contributions
FS, FZM, and MS contributed ideas and participated in writing this article. HRZ contributed to choosing datasets. All authors read and approved the terminal manuscript.
Corresponding author
Ideals declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher'southward Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed nether the terms of the Creative Commons Attribution iv.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided y'all give appropriate credit to the original writer(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Eatables Public Domain Dedication waiver (http://creativecommons.org/publicdomain/nix/1.0/) applies to the data made available in this article, unless otherwise stated.
Reprints and Permissions
Near this commodity

Cite this commodity
Salari, F., Zare-Mirakabad, F., Sadeghi, M. et al. Assessing the impact of exact reads on reducing the error rate of read mapping. BMC Bioinformatics nineteen, 406 (2018). https://doi.org/10.1186/s12859-018-2432-7
-
Received:
-
Accepted:
-
Published:
-
DOI : https://doi.org/10.1186/s12859-018-2432-seven
Keywords
- Reference-based assembly
- Read mapping
- Multi-mapping reads
Source: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2432-7
0 Response to "Samtools Number of Reads Linked to Multiple Contigs"
إرسال تعليق